Global interest and activity in biologics has intensified following the adoption of the Approval Pathway for Biosimilar Biological Products
The Biosimilar Pathway defines a biosimilar as a “biological product [that] is highly similar to the reference product notwithstanding minor differences in clinically inactive components” and requires that there be “no clinically meaningful differences between the biological product in terms of the safety, purity, and potency of the product.”
Now the United States is among many countries in the world having pathways for the approval and marketing of biosimilar, or follow-on biologic, products. It must meet the legal challenges and uncertainties known to other nations in approving biosimilar products. It is not surprising that the Biosimilar Pathway was developed with an understanding that biosimilars are not simply “generic” versions of branded products. More than proof of therapeutic equivalence based on pharmaceutical equivalence and bioequivalence is required for approval of biosimilars.
Biologics are becoming increasingly accessible: According to a recent study, although the biopharmaceutical industry faced significant economic challenges in recent years, sales of “innovator” biologics—that is, a product that is branded, as opposed to generic—rose by 3 percent.
A. An Expedited Licensing Process
One public policy incentive behind the Biosimilar Pathway is to preserve incentives that encourage continued biologics innovation while assuring patient safety and expanding access to life-saving biological therapeutics by maintaining the same fundamental requirements of all biologics sponsors—demonstration of safety, purity, and potency. One function of the Pathway is to utilize all available information on a reference medicine to reduce the amount of information needed to authorize licensing a subsequent biosimilar product. The Pathway established an abbreviated approval regulatory pathway for biological products that are demonstrated to be sufficiently highly similar to, or interchangeable with, an FDA-approved biologic sponsor product. The Pathway, as enacted, provides for up to 12.5 years of non-patent data exclusivity for a biological product approved under a biologics license application (BLA) consisting of a 12-year data exclusivity period that may be extended by meeting additional statutory requirements.
But with speed comes speed bumps. Developing a biologic product, including a biosimilar product, is challenging, and reports have raised concern over the safety of biologics. For example, a 2008 report which took an in-depth look at safety issues surrounding biologics vis-à-vis other types of drugs (i.e., chemical compounds) found that biologics pose a heightened risk of adverse events compared to other types of drugs.
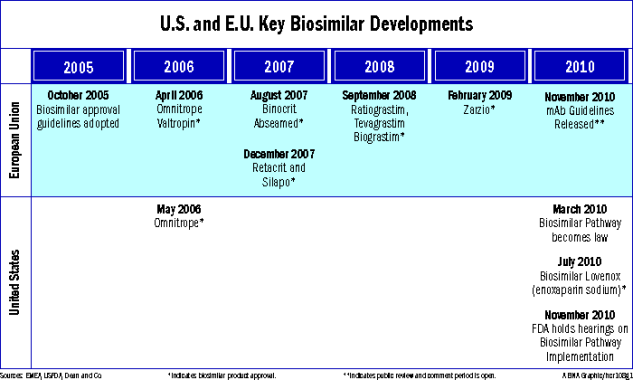
In the United States and Europe, 174 biologics approved between January 1995 and June 2007 were studied. It was found that 41 (23.6 percent) prompted safety-related regulatory actions. Importantly, none of the biologics were withdrawn for safety reasons. But overall, a biologic had a 14 percent chance of prompting safety regulatory action within three years of approval, and a 29 percent chance within 10 years. According to reports, Marisa Papaluca-Amati, deputy head of the European Medicines Agency’s (EMEA) sector for safety and efficacy of medicines, indicated the study was sound but required further evaluation because, for example, there have been and continue to be regulatory changes that should be taken into account.
According to the Pathway, an application submitted for biosimilar approval may not be submitted until four years after the date on which the reference product was first licensed. Other key features of the Pathway include:
- provisions relating to the assessment of biosimilarity;
- a reference product data exclusivity period of at least 12 years after the date on which the reference product was first licensed;
- a first “interchangeable” biosimilar exclusivity period of at least one year; and
- regulations regarding patent listings and alleged patent infringement actions.
B. European and U.S. Regulatory Developments
The EMEA, the European agency responsible for assessing applications from companies to market biological medicines for use in the European Union, including follow-on biologic products, has received requests for at least scientific advice on at least six such antibodies, including one from Teva for a biosimilar version of Rituximab, a genetically engineered chimeric murine/human monoclonal IgG1 kappa antibody directed against the CD20 antigen used to treat, for example, leukemias, transplant rejections, and some autoimmune disorders. According to the EMEA, for biosimilar approval, a company needs to carry out studies to show that the biosimilar is similar to the reference medicine and does not have any meaningful differences in terms of quality, safety or efficacy.
The EMEA continues to monitor the safety of biosimilar medicines once they are on the market. In November 2010, the EMEA released for public comment draft guidelines on biosimilar antibodies (see Timeline). It is likely that the EMEA, at least at the outset, will require substantial clinical trial data using the subject biosimilar antibody in order to recommend authorization of licensure of the biosimilar product; however, the extent of the clinical testing needed to establish biosimilarity in the EU remains uncertain. The EMEA guidelines emphasize early stage comprehensive risk planning, including recommendations to address problems manufacturers may experience during biosimilar monoclonal antibody manufacture (i.e., variability and consistency among assays used to assess antibody immunogenicity).
Also in November 2010, the U.S. Food and Drug Administration held hearings to obtain stakeholder input regarding implementation of the U.S. Biosimilar Pathway generally . Development of biosimilar antibody guidelines in the U.S. will inevitably include debates regarding the standards necessary for obtaining interchangeability approval—that is, the ability to interchange the biosimilar with a sponsor product. To be designated interchangeable, “A sponsor must demonstrate that the biosimilar product can be expected to produce the same clinical result as the reference product in any given patient and, for a biological product that is administered more than once, that the risk of alternating or switching between use of the biosimilar product and the reference product is not greater than the risk of maintaining the patient on the reference product.”
While there are six fusion proteins available in the United States, Enbrel (chimeric recombinant TNF-αreceptor and IgG, developed by Amgen) continues to be the best-selling biologic and Orencia (chimeric CTLA4 extracellular domain and IgG, developed by Bristol-Myers Squibb) is reported to be one of the most rapidly growing fusion protein biologics. These products, among many others, are likely targets for biosimilar development.
C. Trend of Easier `Pathway’ to Approval
Interestingly, according to a recent study conducted by BIO and BioMedTracker, biologics are twice as likely to gain U.S. approval from the FDA than are chemical drugs. Drugs useful for treating infectious and autoimmune diseases are approved more than those for treating cancer. From 2004 through 2010, the overall success rate for the over 4,000 drugs reviewed in the study indicated that drugs moving from early stage Phase I clinical trials to FDA approval is about one in 10, which is substantially less than the one in five to one in six from earlier years. Traditional, small-molecule chemical drugs had a 7 percent success rate while biologics had a 15 percent chance of going from Phase I through to FDA approval.
While a U.S. generic drug pathway existed prior to enactment of the Biosimilar Pathway, a new Pathway was necessary for the approval of biosimilar products because the Hatch-Waxman amendments apply generally to products regulated under the Federal Food, Drug, and Cosmetic Act, not those products regulated under the Public Health Service Act. Because of the fundamental differences between the requirements for generic drugs and biosimilars, the Biosimilar Pathway and the Hatch-Waxman Act should not be confused. For example, the active substance in a drug regulated product can typically be defined, whereas biologics are more complex and heterogeneous. The manufacturing processes for biologics are far more complex and expensive for most biologics, requiring the use of live organisms, which can be difficult to control, than for small molecules.
PHS Act biologics are not listed in the FDA’s Orange Book, such as under Hatch-Waxman, and therefore need to be determined. Included in the patent provisions of the new legislation is a complex default scheme for the confidential exchange of information regarding patent rights relevant to the subject biosimilar and reference (i.e., branded) product. Under the Biosimilar Pathway scheme, after filing of an application with the FDA, the applicant must provide the reference product sponsor (i.e., the innovator) with a complete copy of the biosimilar application, the details of the manufacturing process used in the production of the subject biosimilar and, optionally, any additional information requested by the reference product sponsor. Because of the nature of the disclosures required and the brevity of the statutory time limits set for responding, extreme care should be taken in developing a litigation strategy before filing the application.
D. The Challenging Landscape Ahead
A critical provision of the U.S. Biosimilar Pathway is the 12-year data exclusivity provision, which is now being directly challenged by the administration in the president’s 2012 budget proposal. President Obama aims to slash the period of data exclusivity from 12 years to seven years, thereby attempting to arguably save patient expense by allowing biogenerics to reach the market faster. However, the attack on the law provoked sharp criticism.
Biotechnology Industry Organization President and CEO Jim Greenwood asserted that biotechnology companies would be unable to profit and sustain investments in biosimilars because so much is required to bring such products to market. Regarding data exclusivity, BIO has stated that “biotechnology companies must have some certainty that they can protect their investment in the development of new breakthrough therapies for a substantial period of time in order to secure the necessary resources from venture capital firms and other funding sources” and “the Administration’s budget includes a proposal which flies in the face of President Obama’s own call for the US to ‘win the future’ and maintain our nation’s leadership in research and technology … what is proposed would jeopardize continued biotechnological research and development that will help create new jobs here in the US, lead to new breakthrough cures and therapies, and help us out-innovate the rest of the world . … The biosimilars provision was one of the only bipartisan provisions of the health care law … . Now the Administration is proposing to change this bipartisan, strongly supported provision and provide our innovators with dramatically less protection, in the name of questionable short-term budgetary savings that will come at the price of long-term costs to our healthcare system and our economy.”
Other voices have chimed in. According to industry group Pharmaceutical Research and Manufacturers of America, “PhRMA is disappointed that the President’s budget proposal would diminish crucial incentives for future U.S. medical innovations. … Fair data protection for innovative biologic medicines is critically important to the development of cutting-edge medicines that allow American patients to live longer, healthier and more productive lives. Without such protection, US innovation would be seriously threatened, along with tens of thousands of American jobs” and “…the proposed policy could jeopardize American competitiveness since the US would then provide less data protection for new, innovative biologics than is currently bestowed in Europe.”
It remains unclear what the impact of the administration’s revision of the law will be. It is also uncertain if and when guidelines will be issued by FDA concerning the data necessary to adequately support approval of an application filed under the Biosimilar Pathway. What is sure, however, is that the standards for approval of a biosimilar product will match those of a innovator biologic, namely: safety, purity and potency. FDA has experience in addressing issues related to biologics, such as erythropoietin-stimulating factors, historically, the best-selling category of biologic products.
As regulatory guidelines develop, it is likely that new products will continue to enter the “biosimilar” sector. One example that could be regarded as a biosimilar product is belatacept (the fused Fc fragment of a human IgG1 immunoglobulin linked to the extracellular domain of CTLA-4, a follow-on version of Orencia that differs from Orencia by two amino acids). Belatacept may, at first, face heightened scrutiny, such as the added requirement of submission of data from ongoing trials and/or proposed risk evaluation and mitigation strategy, particularly if safety concerns are raised.
It is also clear that IP management strategies for biosimilars must include an appreciation of the unique and complex nature of biosimilar technologies and comprehensive evaluation of the applicant and sponsor’s intellectual property portfolios.
Learn more about Bloomberg Law or Log In to keep reading:
See Breaking News in Context
Bloomberg Law provides trusted coverage of current events enhanced with legal analysis.
Already a subscriber?
Log in to keep reading or access research tools and resources.