The new EU Biocidal Products Regulation (BPR), EC Regulation 528/2012, repeals and replaces the Biocidal Products Directive (98/8/EC), which regulated biocidal active substances and biocidal products in the EU. The European Commission (EC) clarified many of the BPR’s new provisions to industry in the lead up to adoption of the new regulation on Sept. 1, 2013.
Biocidal active substances and biocidal products are defined as any neat (undiluted or unmixed) substance or product (which may include articles) that exerts an effect on harmful organisms. This “effect” is expansively defined to include anything, including destruction, deterrence, rendering harmless or preventing the action.
Regulation of Biocidal Substances and Biocidal Products
Both the BPD and BPR identify a long list of biocides across 22 “Product Types” (PT) that are produced by a variety of industry sectors and may not be considered “biocidal” in other jurisdictions. The full list of Product Types and their associated descriptions are listed in Table 1 below.
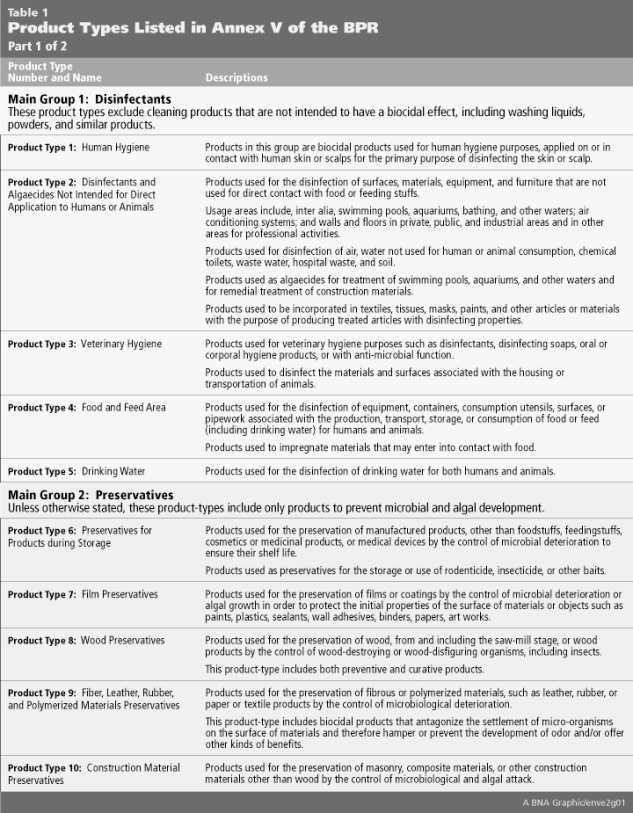
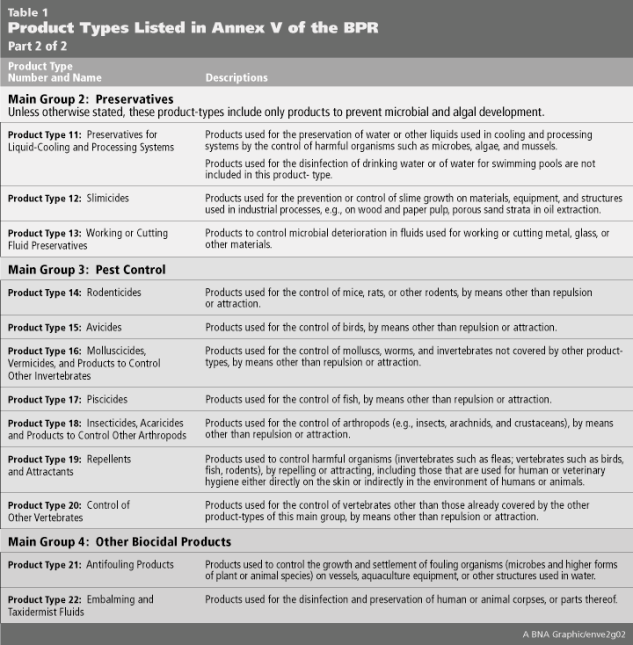
The Product Type 6, “Preservatives for products during storage,” offers a useful illustration of how the BPR differs from biocidal regulations in other jurisdictions. These biocidal active substances are added to products purely for the purpose of ‘in-can’ or ‘in-storage’ preservation, and are often exempted from regulatory control in other jurisdictions. Regulatory disconnects of this type may often present unforeseen challenges and regulatory issues for companies expanding into new territories or product lines. To avoid this type of regulatory pitfall, a search of the Product Type currently approved for the active substance must be undertaken prior to placing the biocidal product on the EU Member State market.
Impact on Biocide Registrants and Downstream Users
Industry sectors across the spectrum will be affected by the BPR due to the scope of the Product Types, including but not limited to the new provisions for “treated articles.” Impacts may be solely financial in nature, where companies will need to obtain authorization to refer to an existing approval or authorization to continue marketing an existing product, or to launch and support a new biocidal product. Some companies may be impacted more extensively, requiring additional resources and the need to address additional administrative burdens. For example, companies wishing to continue importing biocidal products or treated articles that were not previously regulated will need to arrange for authorization of the biocidal product, and in some cases approval of the active substance for the corresponding Product Type for treated articles. This process will take time and money, which must be carefully considered and for which resources will need to be budgeted.
Recognized Suppliers List.
The creation and publication of the “recognized suppliers list” is likely to be the most significant impact on supply chains, both inside and beyond the EU. This list comprises all suppliers of biocidal active substances that have contributed to the cost of the active substance approval and is made publicly available by the European Chemicals Agency (ECHA). Commencing Sept. 1, 2015, only biocidal products containing active substances supplied by a recognized supplier (on the recognized suppliers list) can be legally placed on the EU market.
Technical Equivalence.
Access to an existing approval or authorization (through a “Letter of Access” cost-sharing agreement) is only granted where the identity of the active substance is shown to be “technically equivalent” to the active substance that has already been assessed and approved.
The first tier consists of a basic comparison of the physical and technical parameters of the reference substance to the active substance being assessed. In most cases, the assessment is expected to conclude with a decisive “yes” or “no” conclusion.
The second-tier assessment is conducted when the Tier One evaluation results are inconclusive. This assessment is based on the toxicological and ecotoxicological information available on the reference substance and the active substance to be registered. Testing on vertebrate animals must not be conducted for the sole purpose of technical equivalence.
The technical equivalence assessment is required whenever there is a new source of an approved active ingredient or when the manufacturing process or manufacturing site of the active substance is changed.
Treated Articles.
A new area of regulation covered by the BPR is that of “treated articles.” These are articles such as wooden benches painted with a wood preservative, and clothing treated with biocidal substances to reduce microorganism growth or odor generation.
Companies supplying treated articles to the EU must ensure that all biocidal active substances intentionally incorporated into or used to treat the article, for the purposes of control of harmful organisms, are approved for the relevant PT.
Ensuring Compliance
It is most cost- and time-effective for manufacturers of biocidal products to ensure the biocidal active substances they purchase have already been approved for the relevant Product Type by their suppliers. This is usually viewed as a first step in ensuring compliance and essential to continue a commercial presence on the EU market. Where the biocidal active substance supplier has ensured compliance, a certification or written assurance that all biocidal substances and products are approved or authorized should be provided. If this is not forthcoming, or if the manufacturer/supplier does not possess an approval for the substance and related use, then the biocidal product manufacturer must determine whether to change suppliers to a manufacturer that can provide the approval, pursue the regulatory requirements itself or stop manufacturing such products.
Formulation and manufacturing processes used by manufacturers of biocidal products, in particular biocidal mixtures and preparations used for treatments, are typically considered confidential business information, and manufacturers are therefore reluctant to provide this type of information to their customers. Where this is the case, customers should seek a general certificate of compliance for the product, stating that all biocidal active substances contained in the product are suitably approved. Using this general certification, a supplier may ensure compliance to a customer without stating the specific chemical information. This type of good faith arrangement allows customers to proceed with biocides business and existing supply chains.
Whether a company can reasonably rely on such a certification can depend on several factors (e.g., the size and reputation of the company providing the certification, whether the company expresses a proficiency in BPR requirements, the length and status of the working relationship between the supplier and its customer). Although all entities legally placing an active substance on the EU market will be listed on the recognized suppliers list (as above), this list may not be updated immediately, or the supplier may be using a representative for the purposes of listing. Certification must therefore, always be sought from the supplier as a first step in ensuring regulatory compliance.
To facilitate and expedite inspections by EU Member State agencies, companies must maintain records (electronic or hard copy) of manufacturing processes, quality control and quality assurance for the manufacture of products and batches, and information on safety of products (including any recall notices as applicable). As a minimum, the following must be kept and recorded:
• Safety Data Sheets (SDS) and specification of active substances and other ingredients used in product manufacture;
• Records of substance or product manufacturing operations;
• Results of internal quality control; and
• Identification of production batches.
For manufacturers of active substances or products, retention of samples from production batches also must be kept under appropriate storage conditions.
There is currently a push from ECHA and EU Member State Competent Authorities (under inspection and enforcement activities) for active substances and products to be accompanied by a safety data sheet (SDS) prepared in the format specified in Annex II of the EU Registration, Evaluation, Authorization and Restriction of Chemicals (REACH) regulation (1907/2006), amended by EU regulation 453/2010, and mandated by the BPR.
Enforcement, Penalties for Non-Compliance
Institution of BPR provisions is devolved to local and national agencies in each EU member state, and is enforced under national laws.
In the UK, the BPR provisions are enforced by two agencies, with defined areas of expertise. The Health and Safety Executive (HSE) inspects and enforces any issues arising from the use of biocidal products, such as undisclosed hazards, improper use information or failure to meet approval or authorization provisions in the BPR. The local Trading Standards office, on the other hand, deals primarily with issues relating to retail availability and advertising. The BPR is written into UK national law under the Health and Safety at Work etc Act 1974 (HASWA), in line with the Health and Safety (Enforcing Authority) Regulations 1998 (SI 1998 No 494). Any breach of the BPR in the UK is enforced and prosecuted under HASWA by the HSE.
Maximum penalties are set out in Regulation 32 of The Biocidal Products and Chemicals (Appointment of Authorities and Enforcement) Regulations 2013:
(a) on summary conviction;
(i) in England, Wales and Northern Ireland, imprisonment for a term not exceeding three months or a fine not exceeding the statutory maximum (£5000) or both;
(ii) in Scotland, imprisonment for a term not exceeding 12 months or a fine not exceeding the statutory maximum (£5000) or both; and
(b) on conviction or indictment; imprisonment for a term not exceeding two years, or a fine or both.
Data Compensability Under BPR
Data protection periods under the BPR vary from five to 15 years, depending on the use of the data (for example, new approval/authorization, renewal or amendment). In all cases the trigger for data protection periods is “when [the data] are submitted for the first time.”
The BPR does not include time periods for which data are subject to “exclusive use,” meaning that other interested parties may seek to buy into approvals of active substances, but only after they are made publicly available. This differs from the previous directive and many other regulatory regimes. For example, Section 3(c)(1)(F)(i) of the U.S. Federal Insecticide, Fungicide, and Rodenticide Act (FIFRA) specifies the criteria under which data submitters can receive a 10-year period of exclusive use for certain data submitted in support of a registration for a new pesticide chemical or new uses of an already registered pesticide. Under these provisions, no other registrant is authorized to use the submitted data for the 10-year exclusive use period without the explicit consent of the original registrant, thus effectively creating a private monopoly during the period of exclusivity. This contrasts dramatically with the BPR, where registrants have a mandated duty to share information conducted for the purposes of the active substance approval. The patent-like protection afforded to registrations of new active substances and new biocidal products under FIFRA is missing from the BPR and thus may discourage companies from conducting the necessary research and development activities necessary to keep the EU biocides market innovative and buoyant.
Mandatory Data Sharing, Compensation
The BPR includes a procedure to ensure that companies cannot duplicate tests that have already been performed on vertebrate animals. Instead, any applicant intending to perform a study involving vertebrates must submit a written request to ECHA to determine whether such a study has already been submitted. Although not mandatory, an applicant also may submit a written request to ECHA to determine whether any studies not involving vertebrates have been submitted.
If the data are still protected, the prospective applicant “shall, in the case of data involving tests on vertebrate; and may, in the case of data not involving tests on vertebrates, request from the data owner all the scientific and technical data related to the tests and studies concerned as well as the right to refer to these data when submitting applications.”
The prospective applicant and data owner must make every effort to reach an agreement on the sharing of the results of the tests or studies requested by the prospective applicant.
If an agreement is reached, the data owner will provide the applicant with copies of the data at issue or give permission to refer to the data as set forth in a Letter of Access (LoA). The LoA must include:
(1) The name and contact details of the data owner and beneficiary;
(2) The name of the active substance or biocidal product for which access to the data is authorized;
(3) The date on which the LoA takes effect; and
(4) A list of the submitted data to which the LoA grants citation rights.
If an LoA is later revoked, such revocation shall not affect the validity of the authorization issued on the basis of the LoA in question.
If no agreement is reached, the applicant must so inform ECHA and, within 60 days of being so informed, ECHA can give the applicant permission to refer to the requested vertebrate studies, provided that the applicant demonstrates that every effort has been made to reach an agreement and the prospective applicant has paid the data owner a share of the costs incurred.
This is similar to FIFRA to the extent that the Environmental Protection Agency is likewise allowed to rely upon studies submitted to it even if no compensation agreement has been reached. Compensation for data sharing is to be determined in a fair, transparent, and nondiscriminatory manner. Although the data owner cannot refuse to accept any payment that is offered by the applicant, such acceptance is without prejudice to the data owner’s right to have the proportionate share of the cost determined by a national court. Thus, the BPR provides for national courts, rather than arbitration bodies, to determine compensation when there is a dispute.
As noted above, under the BPR, companies must obtain authorization for the biocidal products and/or approval for the biocidal substances for which they place upon the EU market. Under the prior BPD scheme, companies could market in the EU any active substance or product already approved or authorized, respectively.
The LoA (the document that reflects the permission to refer to a particular dossier and/or specific data referenced therein) is estimated to be significant in cost with a range of several thousands to several tens of thousands Euros. In some exceptional cases (e.g., a small number of data sharers, a high cost for data to support the registration), the cost of the LoA may be much higher, reaching several hundred thousand Euros. The cost is calculated based on the cost of the data incurred to support the authorization or approval, which may include various sources of data, including potential new testing. The data compensation scheme can include additional costs that allow for inflation, risk and administrative fees.
Assistance With Compliance, Registration
Any company (either EU-based or not) that is not familiar with the procedures for biocide registrations, data development or data sharing negotiations, and the administrative burdens placed on industry by the BPR, is likely to contract out compliance and regulatory work to consultants to fulfill their regulatory obligations. Care must be taken in vetting potential service providers to ensure not only a level of knowledge and experience in the EU biocides regulation field, but also a level of scientific competence. The implementation of REACH over the past eight years has seen a boom in consultants offering regulatory services. ECHA does not endorse specific service providers for this type of service, which can make the task of choosing a service provider more difficult.
Future Developments, Amendments
Future amendments are likely to be more in the way of fine-tuning the regulation and tightening up perceived loopholes, contradictions and areas of uncertainty rather than wholesale additions to the regulation. This process has already begun, and proposed amendments to transitional provisions for treated articles and active substances have been developed.
BPR Article 2(5), which exempts food and feed used as attractants or repellents, is expected to be amended to clarify that food or feed used in biocidal products, and sold as an attractant or repellent, is covered by the provisions of the BPR. This amendment would require any food or feed to be approved as an active substance, and listed in the relevant annex of the BPR for the Product Type approved.
The European Economic and Social Committee published the opinion of the Rapporteur Pedro Narro regarding certain conditions of access to the market under the BPR.
Learn more about Bloomberg Law or Log In to keep reading:
See Breaking News in Context
Bloomberg Law provides trusted coverage of current events enhanced with legal analysis.
Already a subscriber?
Log in to keep reading or access research tools and resources.